 |
The first step is to provide the structure of the protein-protein complex :
You may either provide the 4-letter code of the PDB structure, which will then be retrieved from the Protein Data Bank, or upload your own structure file, which must comply with the PDB format. In either case, the structure file should contain at least two distinct protein chains. Please note that: The default structure file on the Protein Data Bank does not necessarily correspond to correct quaternary structure of the protein-protein complex. Select the extension .pdb1, .pdb2, or .pdb3 to use the corresponding Biological Assembly. The format of the "Biological Assembly" files from the Protein Data Bank is somewhat different from the PDB format. In particular, different chains may be referred to as different models of the same chain. In such cases, BeAtMuSiC may have to assign new chain names. In case the structure file contains multiple NMR models, only the first one will be considered.
Once the structure file is uploaded/downloaded, BeAtMuSiC provides the user with a summary of its content. For each chain, the name (if available) and number of residues is given. BeAtMuSiC also identifies sequence-unique entities (groups of chains that share 100% sequence identity, with a coverage of at least 80%).
The second step is to define the two partners of the protein-protein interaction :
Each protein chain present in the structure file must be assigned to either the first or the second partner of the interaction, or be discarded. Both partners must contain at least one chain. Discarded chains will not be taken into account during the computations. 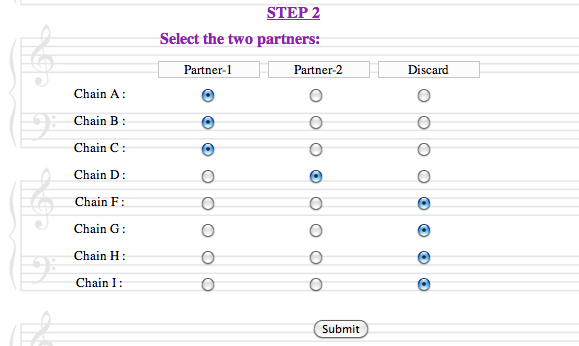
BeAtMuSiC will pre-select two partners, corresponding to the first two sequence-unique entities (or the first two chains in case there is only one sequence-unique entity). This default choice is not necessarily relevant, and the user should be careful in correctly defining the partners of the interactions, since this has a major impact on the computations. - In each partner, as well as in the complex formed by their association, all protein chains should be in contact with each other.
The third step is to select the mutations that will be introduced in the protein chains :
Two options are available: 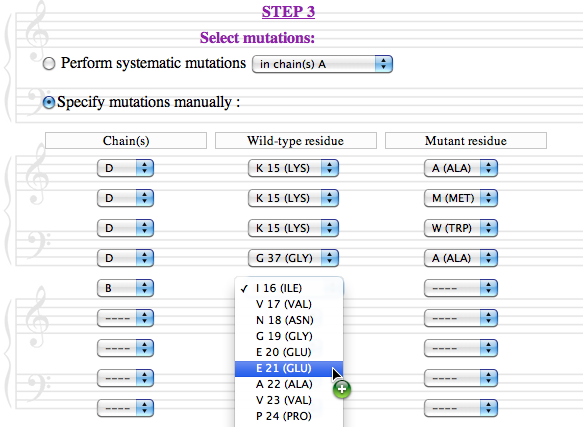
In all cases, sequence-unique entities are treated as groups, and mutations are always introduced simultaneously in all identical chains.
|
 | Upon completion of the third step and submission of the query, BeAtMuSiC provides the "Job ID" and a link that may be followed/bookmarked to check the status of the job. The results will be available on this page as soon as the computations are over (you may need to refresh the page). They will remain on the webserver for two weeks. For each mutation, BeAtMuSiC reports the following information :ΔΔGBind is the predicted change in binding free energy induced by the mutation, in kcal/mol. A negative sign corresponds to a mutation predicted to increase the binding affinity. Please be aware that the reported value corresponds to the total ΔΔGBind resulting from the introduction of the mutation in all identical chains. For example, if the partners are both constituted of two identical chains (eg. AB and CD), the ΔΔGBind value reported will be quite different than if only chains A and C were considered. In the example below, we may expect that ΔΔG1(ABCD) ≈ ΔΔG1(AC) + ΔΔG1(BD) ≈ 2 . ΔΔG1(AC) but, clearly, ΔΔG2(ABCD) ≠ ΔΔG2(AC) + ΔΔG2(BD) since the mutated residue is at the interface between chains A and D, and between chains B and C.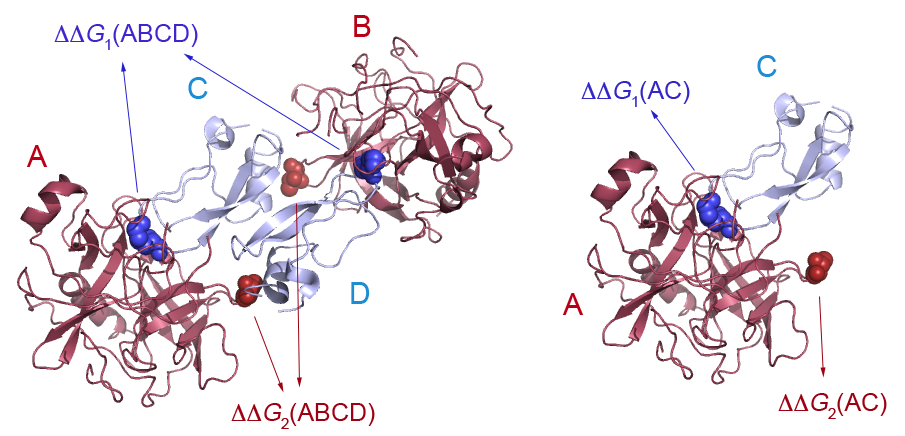
The solvent accessibility of the mutated residue, in the individual partner(s) and in the complex. These values correspond to the ratio of the accessible surface of the residue in the protein and in an extended Gly-X-Gly tripeptide. If the mutation was introduced in several chains of identical sequence, average values are reported.Interface (yes/no) BeAtMuSiC identifies a residue as part of the interface if its solvent accessibility in the complex is at least 5% lower that in the individual partner.
The results may be viewed directly on the browser, or downloaded as a plain text file. If systematic mutations have been performed, the user has the possibility to display the mutations that are predicted to induce the largest increase or decrease in binding affinity, or all mutations at a given position.
|  | In case of systematic mutations in large multimeric assemblies, BeAtMuSiC may take a long time to process the results, or even stall. If the user wishes to evaluate the effect of mutations at an interface that is present multiple times in the structure, it is recommended to select only a subset of the protein chains in order to avoid redundant calculations. The identification of sequence-unique entities relies on the numbering of the amino acids in the structure file. If two chains are identical but numbered differently, they will not be recognized as a sequence-unique entity. In such cases, you may want to edit your structure file and adapt the numbering. This may also be used to introduce a mutation in only one chain and not in other identical chains present in the structure file. In some structure files, chain names are numbers rather than letters. BeAtMuSiC does not always deal correctly with this. Try editing your structure file and changing the chain names. Be aware that non-protein molecules (RNA, DNA, ions, ligands, etc..) are not taken into account during the calculations. Modified residues correctly referenced in the structure file are introduced in the model to preserve chain connectivity, but a larger error should be expected for mutations in their vicinity.
|  | Two different binding models are considered:
In the first model, both partners of the interaction are assumed to be able to fold independently of each other. The change in binding free energy upon mutation is then expressed as follows:
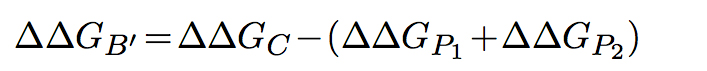 In the second model, the partners are unable to fold independently, and the change in binding free energy upon mutation is thus given by:
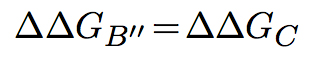
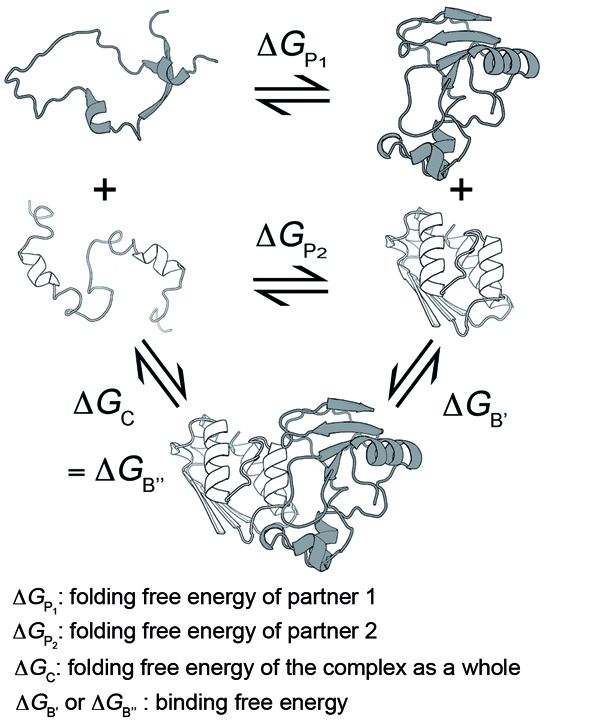
The predictions of BeAtMuSiC are based on the assumption of an intermediate situation, and the change in binding affinity upon mutation is obtained by combining the output of these two binding models:
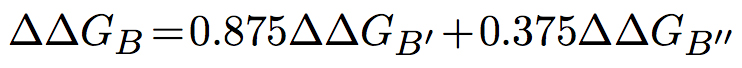 The change in folding free energy of each partner and of the complex, upon mutation, are computed as follows:
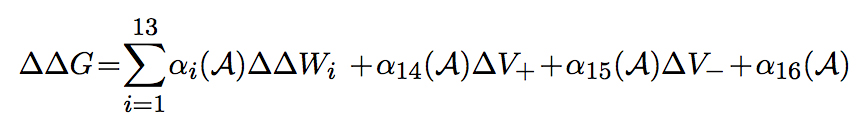
where the weights αi are sigmoid functions of the solvent accessibility, ΔV+ and ΔV- are based on the difference in volume between the wild-type and mutant amino acid, and ΔΔWi corresponds to the energetic change induced by the mutation, according to one of 13 different statistical potentials extracted from a dataset of know protein structures. Local potentials describe the correlations between sequence-structure descriptors associated to residues close to each other along the protein chain:
| ΔWst | 1 amino acid type, 1 domain of backone torsion angles | | ΔWas | 1 amino acid type, 1 domain of solvent accessibility | | ΔWstt | 1 amino acid type, 2 domains of backone torsion angles | | ΔWsst | 2 amino acid types, 1 domain of backone torsion angles | | ΔWaas | 1 amino acid type, 2 domains of solvent accessibility | | ΔWass | 2 amino acid types, 1 domain of solvent accessibility | | ΔWast | 1 amino acid type, 1 domain of backone torsion angles,
1 domain of solvent accessibility |
Distance potentials describe the correlations between sequence-structure descriptors associated to residues separated by a spatial distance d:
| ΔWsd | 1 amino acid type, interresidue distance | | ΔWsds | 2 amino acid types, interresidue distance | | ΔWasd | 1 amino acid type, 1 domain of solvent accessibility, interresidue distance | | ΔWasdas | 2 amino acid types, 2 domains of solvent accessibility, interresidue distance | | ΔWstd | 1 amino acid type, 1 domain of backone torsion angles, interresidue distance | | ΔWstdst | 2 amino acid types, 2 domain of backone torsion angles, interresidue distance |
|  | Don't hesitate to contact us. Feedback is most welcome. - Yves Dehouck: ydehouck@ulb.ac.be
- Jean Marc Kwasigroch: jkwasigr@ulb.ac.be
- Marianne Rooman: mrooman@ulb.ac.be
- Dimitri Gilis: dgilis@ulb.ac.be
|
|
